重症肌无力(myasthenia gravis,MG)是一种主要由乙酰胆碱受体抗体(AChR-Ab)介导、细胞免疫依赖及补体参与,致神经⁃肌肉接头(NMJ)突触后膜信号传递障碍,出现骨骼肌收缩无力的获得性自身免疫性疾病。
MG 全球患病率 150/100万~250/100万,预估年发病率 4/100万~10/100万,没有种族和地区性差异。各个年龄段均可发病,有两个高峰:20~40 岁发病者女性多于男性;40~60 岁男性较多见,多合并胸腺瘤。
MG 的发病机制尚未完全明确,主要与自身抗体介导的突触后膜胆碱酯酶受损有关,胸腺在 MG 的发生发展中起关键作用,胸腺切除手术对改善预后有积极作用和重要意义。
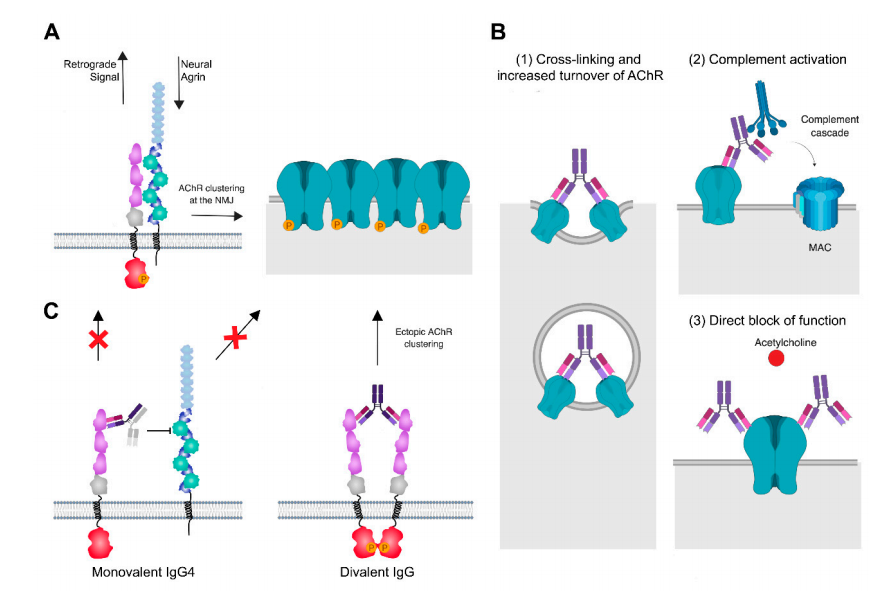
MG 自身抗体在 NMJ 的致病机制[1]
01 临床表现、分型及亚组分类
MG 的主要症状是肌肉无力和疲劳,主要以视物重影、上眼睑下垂为早期表现,逐渐发展为四肢、躯体以及呼吸肌无力,亦有部分以全身症状为起始表现,严重时可危及生命。
MG 少数患者有家族史。常见诱因有感染、手术、精神创伤、全身性疾病、过度疲劳、妊娠、分娩等,有时甚至可以诱发重症肌无力危象。

MG 临床分型丰富,各具特色,以美国重症肌无力基金会(MGFA)临床分型最为细致与直观,MGFA 临床分型、定量 MG 评分(QMGS)项目及评分标准如下表:


MG 临床表现具有极大异质性,以血清抗体及临床特点为基础的亚组分类,对 MG 个体化治疗及预后评估更具指导意义(表3)。
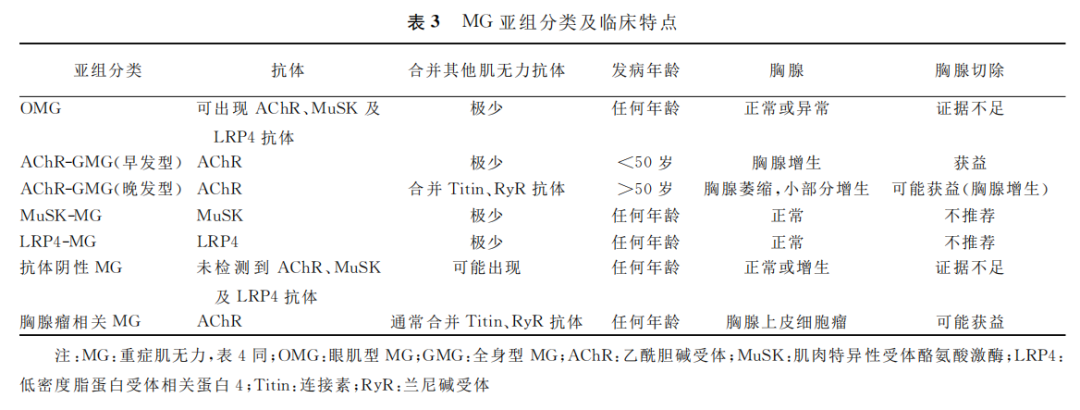
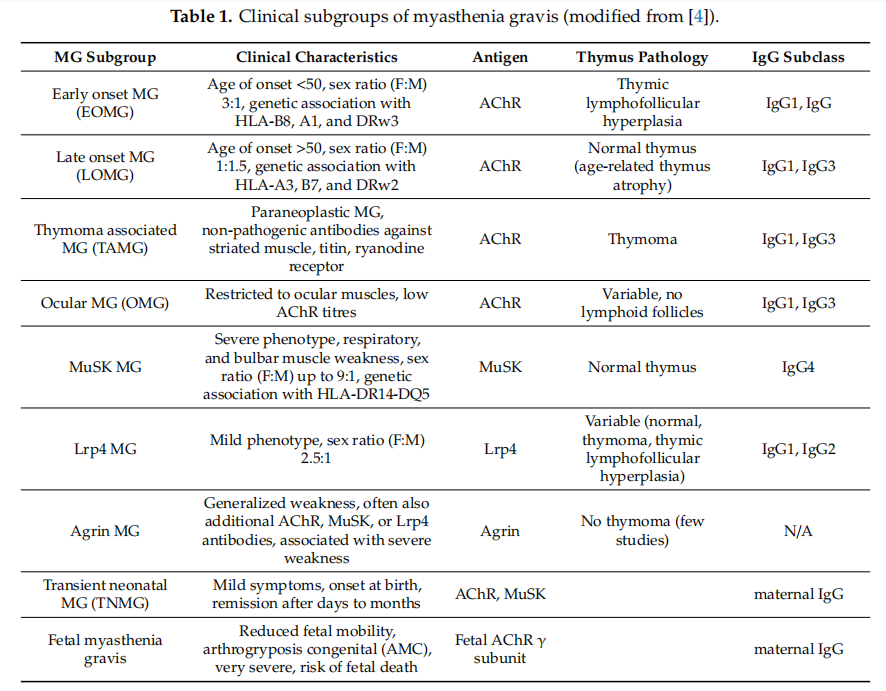
AChR MG 的临床亚组[1]
02 MG 的实验室检查
对于 AChR 和 MuSK 常见的检测方法包括放射免疫沉淀法(RIPA)、酶联免疫吸附测定法(ELISA)和细胞免疫荧光法(CBA),既往研究表明这些检测方法的特异度和敏感度存在客观差异。
MG 自身抗体的实验室诊断推荐[2]:
1. 临床表现高度提示 MG 的患者,在条件允许的情况下建议将 AChR 抗体和 MuSK 抗体检测作为首选辅助检测手段(Ⅰ级推荐,1a 级证据)。
-
约 50%~60% 的 OMG、85%~90% 的 GMG 血清中可检测到 AChR 抗体;10%~20% 的 AChR 抗体阴性 MG 患者血清中可检测到 MuSK 抗体[3]。
2. MG 患者血清 AChR 抗体或 MuSK 抗体的定性分析,建议:
(1)在条件允许的情况下优先考虑高敏感度的 CBA 法;
(2)若诊断方法为 ELISA 或 RIPA,建议对检测结果为阴性的样本用 CBA 进一步验证,或结合临床表现和其他辅助诊断结果进行综合判定。
-
AChR 抗体或 MuSK 抗体的定量分析,建议用 ELISA 或 RIPA 进行抗体定量,用 CBA 进行抗体半定量。
-
AChR 抗体或 MuSK 抗体的定性分析可用于 MG 的诊断和鉴别诊断等,定量分析可用于监测 MG 患者的疾病进展或疗效评估等(Ⅰ级推荐,1b 级证据)。
3. LRP4 抗体在 MG 中的致病性有待进一步研究,LRP4 抗体也常出现于其他疾病如肌萎缩侧索硬化(ALS)。(Ⅰ级推荐,1b 级证据)。
-
在 7%~33% 的 AChR、MuSK 抗体阴性 MG 患者中可检测出 LRP4 抗体[3]。
4. 临床诊断为抗体阴性的 MG 患者,建议间隔6~12 个月或症状进展后进行 MG 相关抗体复测(Ⅱ级推荐,2b 级证据)。
03 诊断与鉴别诊断
1. 与眼肌型 MG(OMG)的鉴别诊断:

2. 与全身型 MG(GMG)的鉴别诊断:

04 治疗措施
MG 治疗的目标是依据 MGFA 对 MG 干预后状态的分级(表4),达到微小状态(MMS)或更好,治疗相关副作用(CTCAE)≤1级。

下面是一些常见的 MG 主要治疗方法:


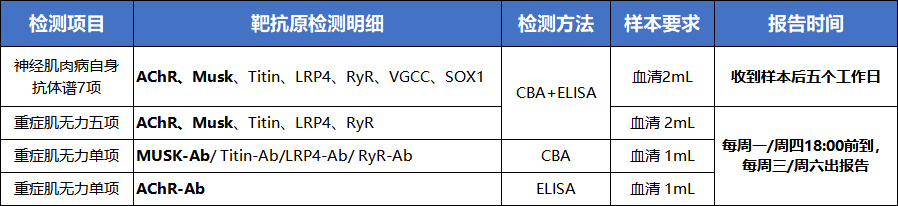
我们提供的重症肌无力检测服务:
重症肌无力相关抗体谱检测:
参考文献:
[1] Inga K, Ruth H. Myasthenia Gravis: Pathogenic Effects of Autoantibodies on Neuromuscular Architecture. Cells, 2019, 8(7): 1-35.
[2] 中华医学会神经病学分会神经免疫学组. 重症肌无力自身抗体实验室诊断专家共识2022 [J] . 中华神经科杂志, 2023, 56(3) : 251-256.
[3] 中国免疫学会神经免疫分会. 中国重症肌无力诊断和治疗指南 (2020 版) [J] . 中国神经免疫学和神经病学杂志, 2021, 28(1) : 1-11.